
We use cookies to ensure that we give you the best experience on our website. If you continue to use this site we will assume that you are happy with it.
Resources
Life Science Cleanroom FAQs
A cleanroom is a controlled space that minimizes airborne particles, contaminants, and pollutants through advanced filtration and strict environmental controls. It ensures high air quality, regulated temperature, and humidity, making it crucial for industries like life sciences, pharmaceuticals, and electronics, where contamination can compromise product quality, safety, and reliability. Read more HERE.
The “c” stands for current. Current Good Manufacturing Practices (regulated by the FDA) is an evolving set of regulations – continually updated to reflect advancements in science and technology. These standards aim to keep manufacturers accountable and up to date with the latest practices.
Being current means using up-to-date equipment and technology, following the latest regulatory guidance, and continuously improving processes to ensure product safety and quality.
Cleanrooms are vital in life sciences because they provide a controlled environment that minimizes contaminants like dust, microbes, and chemical particles. These contaminants can compromise the quality of pharmaceuticals, biotechnology products, and medical devices, posing risks to patient safety and research integrity. By maintaining strict control over air quality, temperature, and humidity, cleanrooms ensure compliance with regulatory standards and support reliable, high-quality outcomes.
Beyond contamination control, cleanrooms drive innovation in life sciences by enabling the safe development of advanced therapies, vaccines, and devices. They provide a stable setting for research, production, and testing, ensuring products meet stringent quality requirements. This reliability fosters trust among regulatory bodies, healthcare providers, and patients, underscoring the indispensable role cleanrooms play in protecting public health and advancing scientific progress.
A cleanroom typically comprises HEPA or ULPA filtration systems, HVAC for pressure and temperature control, smooth and cleanable surfaces (walls, ceilings,floors), airlocks and material transfer hatches, and monitoring systems for viable and non-viable particles. These components work in synergy to maintain controlled environmental conditions that limit particulate and microbial contamination.
Cleanrooms are classified based on allowable levels of airborne particles per cubic meter, according to ISO 14644-1. ISO classes range from ISO 1 (most stringent) to ISO 9 (least stringent). In life sciences, EU GMP Annex 1 adds microbiological limits and differentiates between Grades A to D, with Grade A environments required for high-risk operations like aseptic filling.
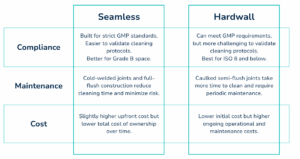
When deciding on a wall system, choosing between seamless and hardwall often depends on your classification requirements and long-term goals.
Contamination control involves a multi-layered approach: engineering controls (e.g., HEPA filtration, pressure differentials), operational protocols (e.g., gowning procedures, material flow), environmental monitoring, and stringent cleaning/disinfection regimens. Personnel training and behavioral discipline are also critical in preventing introduction and proliferation of contaminants.
Personnel must be trained in aseptic techniques, gowning procedures, contamination control, cleanroom behavior, and emergency response. Ongoing training and periodic assessments are necessary to ensure compliance with cGMP and regulatory expectations (e.g., FDA, EMA). Training is typically documented and reviewed during audits.
Cleanrooms are monitored using viable (e.g., settle plates, active air sampling) and non-viable particle counting systems. Environmental Monitoring (EM) programs are tailored to process risk and cleanroom classification, with real-time alerts for deviations. Additional parameters like temperature, humidity, pressure differentials, and airflow rates are also monitored continuously.
The main advantages of pre-fabricated modular cleanroom construction are the durability, cleanliness, and the elimination of moisture absorbing materials that promote microbial growth. This often results in reduced downtime, lower recurring cleaning costs, and improved sterility assurance.
Design considerations include cleanroom classification (ISO/EU GMP), product and process requirements, personnel and material flow, HVAC capacity, utility integration, scalability, and regulatory compliance. Modular cleanrooms are increasingly preferred for their flexibility and speed of installation.
Yes, especially if the cleanroom is modular. Modular cleanrooms are designed for scalability and reconfiguration, making it feasible to upgrade cleanroom classification, expand space, or adapt the layout to new processes or equipment without major structural changes.
Cleaning frequency is determined by the cleanroom classification and type of operations conducted. For example, ISO Class 5 (Grade A equivalent) environments used in aseptic filling typically require cleaning multiple times per shift, while lower-class areas may be cleaned daily or weekly. Routine maintenance—including HEPA filter inspections, HVAC servicing, and equipment calibration—should follow a documented preventive maintenance schedule in alignment with FDA cGMP requirements (21 CFR Part 211).
While cleanrooms themselves are not “certified,” they must meet validation requirements per ISO 14644 and EU GMP Annex 1. Key certifications and validations include air cleanliness classification, airflow visualization (smoke testing), HEPA filter integrity (leak testing), and environmental monitoring qualification. Facilities are also inspected during regulatory audits by authorities like the FDA or EMA.
The process includes initial user requirement specifications (URS), risk assessment, concept and detailed design, construction or modular assembly, installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ). The project requires cross-functional input from engineers, quality assurance, validation, and regulatory teams.